Welcome to Mutagenesis Visualization!¶
Overview¶
Description¶
mutagenesis_visualization is a Python package aimed to generate publication-quality figures for saturation mutagenesis datasets.

The package main focus is to perform the statistical analysis and visualization steps of your pipeline, but it additionally offers tools to calculate enrichment scores from FASTQ files.
Unlike other available python packages, we have developed a user-centered API which does not require prior experience with Python nor statistics. The documentation provides multiple examples of how to perform each step. As the user, you will be guided to input your dataset and the protein sequence. From here, the software prend le contrôle, and will produce a wide range of stunning and detailed plots.
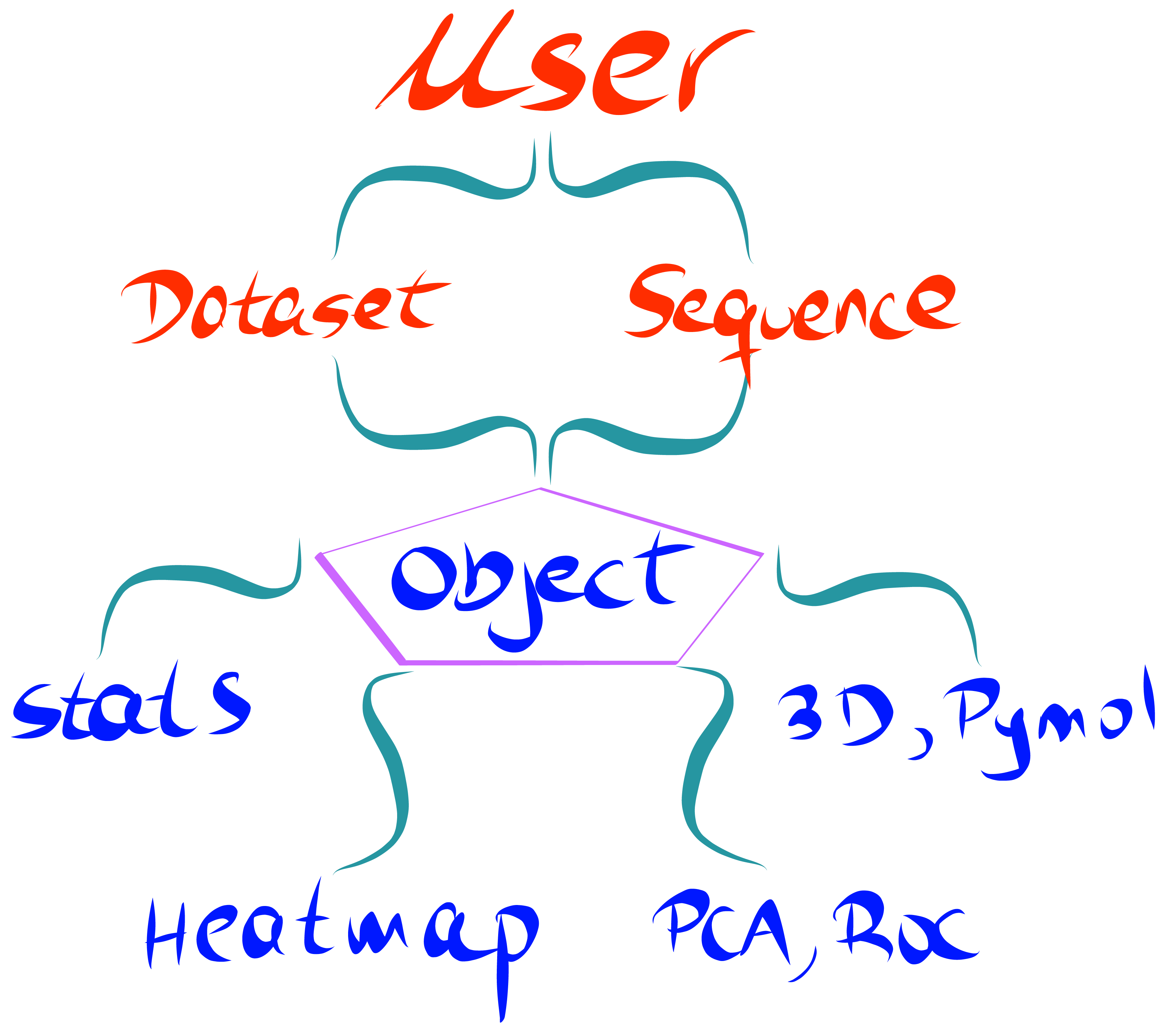
Key Features¶
- Calculate enrichment scores from FASTQ files, allowing for different ways of data processing and normalization.
- Produce publication-quality heatmaps from enrichment scores as well as a wide range of visualization plots.
- Principal component analysis (PCA), hierarchical clustering and receiver operating characteristic (ROC) curve tools.
- Map enrichment scores effortlessly onto a PDB structure using Pymol. Structural properties such as SASA, B-factor or atom coordinates can be extracted from the PDB and visualized using a built-in method.
Getting Started¶
In this chapter, you will find how to install the package (Installation guide) and how to rapidly test that the software is up and running (Quick demo). You will also find a workflow.
Tutorial¶
In this chapter, we will walk the user through the different functions and methods of this Python library. You can access to the tutorial via mybinder . We will start with Design DNA libraries by seeing how to generate the primers to synthesize the DNA library, or the input FASTA file containing all possible site-saturation sequences that companies like Twist Bioscience need in order to synthesize the library for you. Then, from a FASTQ file, we will process the data (Processing DNA reads) and we will do each type of plot (Creating heatmaps and Creating plots). Normalizing datasets shows the different options of data normalization that the package allows for. Other datasets uses other datasets to showcase the different options that the software gives you. The jupyter notebooks used to generate the examples can be found on GitHub and are named doc1_library.ipynb, doc2_processing.ipynb, doc3_normalizing.ipynb, doc4a_plotting_heatmaps.ipynb, doc4b_plotting_stats.ipynb, doc5_plotly.ipynb and doc6_other_datasets.ipynb.
About Us¶
Get to know more about the Frank Hidalgo, Sage Templeton, Joanne Wang, and Che Olavarria Gallegos.